
Northwest Biotherapeutics Update - published 15th May 2022
As I'm sure you are well aware, NWBO finally released the topline results for their phase 3 trial of DCVax-L in newly diagnosed glioblastoma that we have all been eagerly anticipating. Therefore, it was clearly time for an update on my analysis and opinion on this new information since I've now had a couple of days to digest the information.
Due to illness Dr. Paul Mulholland presented the data instead of Dr Linda Liau. The slides were published concurrently with the presentation and they can be found here:
https://virtualtrials.org/dcvax.cfm
It was announced that the primary endpoint of OS in ndGBM was met with a hazard ratio of 0.80 and a p value <0.002. The secondary endpoint of OS in rGBM was met with a hazard ratio of 0.58 and a p value <0.001. The secondary endpoint of PFS (original primary endpoint) failed with HR of 1.1 and p value 0.47. No data was presented on the other secondary endpoints; secondary OS of DCvax-L/SOC arm versus SOC control arm, confirmed PFS of DCVax-L/SOC arm versus SOC control arm and tumor response of DCVax-L/SOC arm versus SOC arm.
So overall I was slightly disappointed with the results. I was hoping the original ndGBM OS endpoint of DCVax-L/SOC versus SOC alone had hit as suggested as a possibility by my previous simulations from February 2020 (analysis here). In my opinion the smart money was looking past the endpoints comparing the historical controls to the original ndGBM OS DCVax-L/SOC versus SOC readout. The results of this endpoint were not announced in the presentation which suggests it may have failed. However, to delve deeper we can do 2 things:
- Look at the mOS data from slide 29 green line and compare that with the mOS from blended results from liau 2018. In the final data presented here the mOS is 22.4 months from surgery whilst in Liau et al., 2018 the mOS number was 23.1 months. That means that by removing the control patients from the 2018 blended data results in the mOS decreasing. This suggests that the SOC concurrent control patients actually performed better than the DCVax-L/SOC patients in the trial suggesting the endpoint failed.
- mOS is only a single point on the KM graph however. A more precise way to get an idea of what is happening is to overlay the graph from slide 29 green line with the blended results from liau 2018. When we do that we see the following:
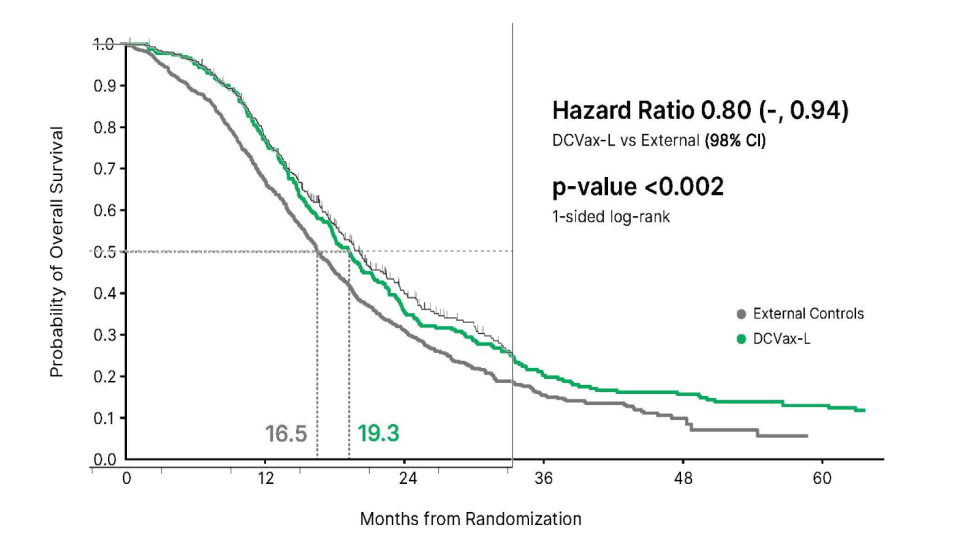
The blended data from liau 2018 can be seen as the thin black line with censor marks on top. I purposely left some of the features (median dashed line and x-axis) from the blended data so you can judge my alignment for yourself. The blended data is shifted over 3 months to take account of the fact that it is measured from surgery whilst the new published final P3 data is from enrollment. As you can see the curves are almost identical with the blended slightly outperforming the DCVax-L final data. This strongly suggests there was no difference in OS between DCVax-L/SOC and SOC arms.
To be clear these overlaid graphs tell us nothing about the tail, so in theory the endpoint could be positive if the survival curves diverge later in time >36 months, but this seems unlikely given the size of the divergence required. This likely failure of DCVax-L/SOC to improve OS compared to SOC alone is why I think there was a big drop in the NWBO share price throughout the morning of the presentation. I don’t think this had anything to do with the original PFS primary endpoint failing as I think almost everyone expected this to fail due to well documented pseudoprogression in immuno-oncology trials.
So there appears to be no difference in the outcome depending on which arm of the trial the patients were enrolled. However, patients from the trial (in either DCVax-L/SOC or SOC arm) genuinely seem to be outperforming those from the historical control cohort as can be seen by comparing the survival curves for the blended cohort (thin black line) and DCVax-L/SOC arm (green line) to the historical control cohort (grey line) in the overlaid figure i just showed. The question thus shifts to why all of the patients in the DCVax-L P3 trial did so well? There are at least 3 possibilities that could explain this 1) chance bias creating a better than expected prognosis for the DCVax-L P3 patient cohort, 2) selection bias due to the inclusion/exclusion criteria creating a better prognosis for patients in the DCVax-L P3 cohort and 3) That DCVax-L has efficacy for both ndGBM and rGBM since due to cross-over approximately 90% of the patients in the DCVax-L P3 cohort were treated with DCVax-L at some point in the trial.
Though the company tried to as equally as possible match the patient cohort from the DCVax-L P3 to historical controls by using an independent third party to construct the data set, there are still some deviations in the patient characteristics. There are two features that may have slightly favoured the prognosis for patients in the DCVax-L P3 trial compared to historical controls:
- Mainly by chance (since most trials for ndGBM require a good KPS score to enrol), the DCVax-L trial had slightly healthier patients (69% >KPS 90) compared to the historical controls used for this study (64% >KPS 90)
- Most likely entirely by chance the DCVax-L trial had slightly more of the better prognosis MGMT methylated patients (39% MGMT+) compared to the historical controls used for this study (32% MGMT+).
Overall, I do not think these two features alone are enough to explain the difference in outcome seen between the DCVax-L patient cohort and the historical controls. In particular the survival at 60 months of 13% presented for the DCVax-L/SOC 232 patient cohort would be difficult for these features to account for especially considering the length of follow up in the DCVax-L P3 trial (meaning that its 60 month readout is likely accurate).
The main selection bias that could result from the inclusion/exclusion criteria used in the DCVax-L P3 trial that could create a false positive is the ‘intent for gross resection’. Obviously gross resection is required in order to get enough tissue to make sufficient vaccine doses hence why there is this requirement to enrol in the trial.
The company published some KM analysis comparing subsets of the patients from the DCVax-L/SOC arm to historical controls. Though these analyses are only exploratory since the trial is not powered to explore such multiplicity they are useful to look at in borderline approval situations such as this one. In particular in the slides that the company published, they showed the KM data for minimal residual disease, which showed that surprisingly only patients with significant residual disease beat the historical controls, whilst patients with minimal residual disease performed equally well in both the DCVax-L/SOC and historical control arms. At first viewing this might suggest that ‘intent for gross resection’ therefore can’t be the explanation for why the concurrent control SOC arm in the P3 performed so well. However, residual disease was not what was being selected for in this trial. They should have shown the results for gross resection versus partial resection which was actually what they selected for in this trial. For example a patient could have had significant residual disease yet also had gross resection. This is one of the first pieces of data that I would expect an examiner for the FDA/EMA would want to see from the full dataset.
However, even if I don't think the most appropriate data has been shown, I still doubt that degree of resection is the explanation as to why the patients fared better in the DCVax-L P3 trial compared to historical controls. The reason for this is the original analysis that i did back in February 2020 where i compared the blended data from Liau et al., 2018 to trials with similar levels of resection and that demonstrated that the DCVax-L data typically improved survival in a statistically significant fashion (analysis here).
The last possibility in this list is that DCVax-L genuinely has efficacy for both ndGBM and rGBM. Due to enrollment and ethical reasons a cross-over option was required for patients in the DCVax-L P3 trial that originally started on the SOC arm to switch to the DCVax-L/SOC arm upon recurrence. Because of this cross-over option, 90% of the patients in the trial took DCVax-L at some point (232 from the beginning and 64 upon recurrence). This is the reason that NWBO gives as to why they changed the DCVax-L trial to principally a single arm trial comparing the DCVAx-L/SOC arm to historical controls. The company also used the opportunity to create a new secondary endpoint for recurrent GBM comparing those patients that crossed over from SOC to DCVax-L/SOC to historical controls for rGBM. If DCVax-L has efficacy for both ndGBM and rGBM then we would expect the patient cohort in the DCVax-L P3 to outperform historical controls because nearly all (90%) of the patients in a ndGBM (DCVax-L/SOC arm) or rGBM (SOC>DCVax-L/SOC cross-overs) setting had taken DCVax-L at some point.
In the ndGBM setting patients in the DCVax-L P3 on the DCVax-L/SOC arm clearly outperform historical controls as shown by slide 29 (HR=0.8 with p<0.002). The question is then the SOC arm of the DCVax-L P3 trial. The data presented in slide 39 shows the SOC>DCVax-L/SOC cross-over cohort beating historical controls with an HR of 0.58 and p-value of <0.001. At first sight this seems very impressive, but there is a major problem with cross-over bias here that should have been addressed.
Cross over patients almost always have a better prognosis than non-cross over patients, simply because a patient needs to be healthy enough to choose to cross over and undergo further treatment. In order to judge whether DCVax-L really does improve the prognosis in rGBM it is necessary to include the 30 or so patient cohort that had tumour recurrence but did not receive DCVax-L (even if this will dilute any genuine efficacy signal by means of adding patients that never received DCVax-L). Previously, I have stated that this rGBM OS endpoint was just a long shot for the company to get approval for rGBM aswell, but If the resulting graph indeed shows that the full DCVax-L/SOC concurrent control arm (n=99) performs better than the historical controls anywhere similar to the effect size seen here (HR=0.58 with p<0.001) then I believe this is good evidence of efficacy of DCVax-L for ndGBM and rGBM and should to support the approval of both.
Finally, there were 3 other secondary endpoints in this trial that were never mentioned in the presented results; OS of DCvax-L/SOC arm versus SOC control arm, confirmed PFS of DCVax-L/SOC arm versus SOC control arm and tumor response of DCVax-L/SOC arm versus SOC arm. Were they not mentioned as they were not met or for some other reason? I have already discussed the original OS endpoint between DCVax-L/SOC and SOC that has been changed to a new secondary endpoint. For that endpoint we can reverse engineer a reasonably good guess of what it looks like. Of the other endpoints meeting the endpoint for Confirmed PFS would be another major piece of evidence that would support approval depending on the exact effect size/p-value. However, PFS is typically a pseudo endpoint for OS simply used to get to a readout faster and given the length of follow-up the company now has for OS this data is not the most important.
Overall I feel I am lacking enough information to take a strong opinion on the functionality of DCVax-L and likelihood of approval given the new data presented. The 3 pieces of data i would like to see include 1) the KM analysis of historical rGBM controls versus the full DCVAx-L placebo arm (99 patients) since recurrence. 2) the KM analysis of the test DCVax-L group versus the ndGBM historical controls when stratified for gross versus partial resection and 3) The confirmed PFS endpoint and KM analysis between the DCVax-L/SOC and SOC arms of the trial. I hope that a publication is coming soon that contains these details to help generate a more informed opinion.
While the original OS endpoint result (DCVax-L/SOC versus SOC) is likely to be disappointing, there is clearly a divergence here between what is financially better for me personally and for scientific purity (i.e. stringently concurrently controlled data sets) versus what is better for patients. If it indeed turns out that the reason the original OS endpoint endpoint (DCVax-L/SOC versus SOC) fails is because DCVax-L indeed has efficacy in rGBM and the SOC>DCVax-L/SOC cross-overs simply diluted any signal that could have been measured then this is positive in multiple ways 1) it would that be great for rGBM patients in general and 2) it would have been particularly good for those patients in the DCVax-L P3 that started on the SOC arm but ended up crossing over to the DCVax-L/SOC arm since they would have given themselves the greatest chance of living longer.
Finally, if the effect size for rGBM OS is as big for the whole SOC arm cohort as the company is currently showing for the SOC>DCVax-L/SOC cohort (HR=0.58 with p<0.001) and that can be shown not to be due to the 'intent to gross resect' inclusion/exclusion criteria then in my opinion DCVax-L should be approved for both ndGBM and rGBM. Approval is always based on a tradeoff between patient benefit and side effects and given the minimal amount of SAEs that could potentially be due to DCVax-L any hint of patient benefit should result in approval. At worst I would like to think that the EMA/FDA would conditionally approve DCVax-L for both ndGBM and rGBM but with the need to perform confirmatory trials with proper concurrent controls (if they are ethically and practically feasible) to confirm efficacy.
I remain long NWBO and my position has not changed.
See terms of use tab.
Northwest Biotherapeutics Update - published 23rd July 2021
Northwest Biotherapeutics (NWBO) is currently my largest investment. NWBO is developing a treatment called DCVax-L for the treatment of newly diagnosed Glioblastoma which is currently in a phase III international drug trial.
It’s been over a year now since I wrote the NWBO piece (that can be found below this update) and there have been some important changes since then which I thought were overdue for a small update. First I will discuss some previous analysis before discussing the significance of the endpoint changes that were announced on the British and European EU clinical trials sites in October of 2020. Finally, I will discuss my expectations regarding the results of the trial.
One oversight on my part was that the measurements I am using are likely to overestimate the p-value of a distinction between the DCVax-L+SOC and SOC alone arms (presuming there is a distinction - and by that I mean the p-value should come out lower than predicted from my previous analysis). The reason for this is that the blended data presented so far by the company (Liau et al., 2018) was only presented up to 36 months. The trial has much more mature data than this, in theory upto over 12 years since the first patient was enrolled in June 2008 and data was confirmed locked in October 2020. Though it is unlikely they have KM data for this long as the KM data for analysis purposes can only run until one of the arms has fully evented which is likely to occur much earlier than 12 years (but much longer than 36 months).
Related to this oversight, it is clearly not possible to do the Fleming-Harrington 0-1 analysis that I was planning since we do not have the data at the right-hand side of the KM graph to perform such an analysis. Overall however, my belief that DCVax-L is functional for improving survival of newly diagnosed Glioblastoma patients has increased due to this overestimation.
The endpoints for the trial were recently updated in October 2020 for the British and German versions of the European trials database. The British and German versions can be found here respectively:
https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-001977-13/GB
https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-001977-13/DE
The endpoints consist of a single primary endpoint and 5 secondary endpoints as follows:
-
The primary endpoint of this study is overall survival (OS) compared between patients randomized to DCVax-L and control patients from comparable, contemporaneous trials who received standard of care therapy only, in patients with newly diagnosed glioblastoma.
-
The first secondary endpoint is overall survival (OS) compared between patients randomized to placebo who received DCVax-L treatment following disease recurrence, and control patients from comparable, contemporaneous clinical trials, in patients with recurrent GBM.
-
The second secondary endpoint, confirmed progression-free survival (cPFS), is confirmed disease progression (cPD) compared between subjects randomized to DCVax®-L and those randomized to Placebo within Study 020221.
-
The third secondary endpoint, PFS, is progression-free survival compared between subjects randomized to DCVax®-L and those randomized to Placebo within Study 020221.
-
The fourth secondary objective, OS, is overall survival compared between subjects randomized to DCVax®-L and those randomized to Placebo within Study 020221.
-
The fifth secondary objective is tumor response compared between subjects randomized to DCVax®-L and those randomized to Placebo within Study 020221.
To summarize, the new primary endpoint is a comparison of overall survival of all patients from the trial (DCVax-L combined with DCVax-L+SOC) and comparable patients from historical trials effectively turning the trial into a single arm trial. The new first secondary endpoint suggests the company is looking at the possibility of getting DCVAx-L+SOC also approved to treat recurrent GBM since approximately 90% of those control patients crossed over to the DCVax-L treatment upon disease progression. The new second secondary endpoint is the improved version of the previous primary PFS endpoint where progression must first be confirmed. There was clearly a problem with pseudoprogression in this trial which needed to be rectified and this endpoint is intended to do that. The third and fourth secondary endpoints are the same as the original primary (PFS) and secondary (OS) endpoints of the trial.
My expectations regarding the new primary endpoint depends on what trials and patient subsets the company has chosen for the historical comparison. I have seen many commentators comparing the DCVax-L PIII to the full datasets from other recent trials in ndGBM such as the optune PIII trial (Stupp et al., 2017) or the Rindopepimut PIII trial (Weller et al., 2017) to name but a few. If that is the case then personally I don't even think we need to see the results to know that the DCVAx-L P3 trial has met its new primary endpoint of bettering historical controls for OS. The blended data from liau 2018 is enough to determine this since one can already compare the data. The difference is stark enough between Liau 2018 and most of the historical data that in my opinion an analytical reconstruction is really not necessary to determine the result will be statistically significant.
However, this is not what I hope the company has done. My hope is that the contemporary patient subset which is compared to the DCVax-L P3 data has been drawn up in an unbiased fashion by an independent set of experts who have attempted to as closely as possible match the patient characteristics between those trials and the DCVax-L P3 if they could access the individual patient files from other trials. Getting hold of individual patient files from past trials may not be so easy however for various reasons including data exclusivity. If this is indeed what the company has done then I would expect the result to look something akin to the markov model in the analysis tab shown in this webpage (analysis link). Again I would expect the trial to be significant given the Markov analysis and given that 1) the data is now much more than the interim data used for the Markov analysis performed here and 2) We have not included the >36 months survival contribution to the statistic.
Regarding the new secondary endpoint. To me this seems like a hopeful long shot to get approval also for recurrent GBM since they are aiming to compare the crossover patients from the placebo SOC arm of the DCVax-L PIII to historical data. Cross-over patients almost always fare better than the cohort from which they derived since one must be healthy enough to choose to cross over. As such they are normally biased groups of patients and I doubt this would fly with the regulatory authorities. I would expect another phase 3 trial to be necessary for NWBO to get approval for DCVax-L to treat newly diagnosed Glioblastoma.
The new second secondary endpoint strongly suggests that they indeed had pseudoprogression issues with the trial. Success with this endpoint could provide a lot of weight for getting approval since this endpoint is a direct comparison between the treatment arm and concurrent control. The same goes for the new fifth secondary endpoint of objective response (Tumour shrinkage) compared between concurrent treatment and control arms. The new 3rd and 4th secondary endpoints are the original endpoints comparing PFS and OS between concurrent treatment and control arms. Pseudoprogression and cross over are likely to have caused problems with these endpoints respectively but I think OS in particular still has a chance of coming out statistically significant. In my opinion these original endpoints will still carry the bulk of the weight of evidence considered when it comes to approval by the regulatory agencies.
To summarize, without seeing the Statistical Analysis Plan it is impossible to tell exactly what historical data the DCVax-L trial data will be compared to, what analysis they will use and what is the breakdown of the alpha expenditure in these endpoints. The EU website reads “This endpoint will be assessed using 3 different analyses”. A slide recently used in one of Linda Liau’s talks suggests these could be Cox proportional hazards, Conditional survival and Subgroups/Cox PH for the primary endpoint but again this has not been confirmed to be the analysis that the company will use. Overall approval of the drug will be decided by the entirety of the evidence and I am still hopeful that NWBO will have enough evidence to get approval. Based on my original Markov modelling (analysis link) I fundamentally believe that DCVax-L is functional for newly diagnosed Glioblastoma and one or more of the new endpoints will show it.
Finally, the company has stated that they are waiting to produce a publication to release topline data. I expect this is because the company needs to explain in detail why those particular contemporary trials and patient subsets have been chosen for comparison in the primary endpoint, what analysis has been done and how the alpha has been spent through those analyses. This trial is the most important trial in Glioblastoma for approximately 2 decades. As such it will very likely be published in NEJM or the Lancet so long as it includes the data for all endpoints and what is a formal pass and what is purely exploratory is clearly spelled out as I expect it will be. Just like everyone else I look forward to seeing the data and the article. I remain long NWBO.
See terms of use tab.
Northwest Biotherapeutics - published 28th February 2020
Northwest Biotherapeutics (NWBO) is currently my largest investment. NWBO is developing a treatment called DCVax-L for the treatment of newly diagnosed Glioblastoma which is currently in a phase III international drug trial.
Glioblastoma is also known as Glioblastoma Malforme is the most aggressive brain cancer that originates from the brain. The prognosis of the disease is poor with fewer than 3-5% of patients surviving 5 years. The current standard of care in the Stupps protocol which involves chemoradiotherapy. According to the original study (Stupp et al., 2005) the protocol involves 2Gy of radiotherapy for 5 day bouts over the space of 6 weeks with concurrent temozolomide 75mg/m2 of body surface area per day for 7 days a week. After this maintenance temozolomide is administered consisting of 150-200mg/m2 for 5 days during 28-day cycles.
DCVax-L is a cancer vaccine based on dendritic cells which has been developed over the last couple of decades by Linda Liau and colleagues at UCLA and is currently being commercialized by Northwest Biotherapeutics. Dendritic cells are the master antigen presenter of the immune system presenting sampled antigens from the surrounding environment to T cells to coordinate an immune reaction to non-self. DCVax-L involves resection of a solid tumour followed by the generation of a tumour lysate by a combination of mincing, enzymatic treatment, freeze-thawing and centrifugation. Dendritic cells are purified from the patient following leukapherisis. The dendritic cells are then grown in culture and pulsed with the lysate and appropriate cytokines. Those Dendritic cells are then reinjected back into the patient intradermally with a specific dosing schedule once they have finished a course of chemoradiation with a specific dosing schedule with concurrent chemotherapy. The mechanism of action has been suggested to be an adaptive immune response directed against the tumour by the dendritic cells primed by the tumour lysate.
The attraction of such a therapy is obvious since it is more of a platform technology than a single therapy as the same pipeline can theoretically be used on any type of solid tumour. Demonstration of efficacy in one tumour setting (and especially one of the hardest tumours to treat) partially validates the treatment for other solid tumour settings.
The current phase III follows the gold standard set-up for a clinical trial; multicenter, international, double-blinded and placebo controlled. It started recruiting in 2007, though it had a pause of approximately 2 years due to the financial crisis. Recruitment terminated in November of 2015. Originally it was intended as a phase II study but was rescaled to a full phase 3 study. The company published blended blinded data of overall survival in May 2018 (Liau et al., 2018). In total 331 patients were recruited, 99 to the Standard of Care (SOC) control arm and 232 patients to the treatment+SOC arm. The blended median survival for this blended population was 23.1 months. The survival data has since been updated but it is not formally publically available. The original set-up was a primary endpoint of PFS and secondary of OS.
DCVax-L originally went through 2 early phase I trials (Liau et al., 2005; Prins et al., 2011). In Liau et al., 2005 was a phase I study where seven patients had newly diagnosed tumors, whereas five had recurrent glioblastoma multiforme to make a total of 12 patients. This study was obviously single arm and open label and was compared to patients treated historically/concurrently with SOC. Median survival was 23.4 months for the full group compared to 18.3 months for SOC. For the patients with stable tumors or no residual disease at the time of dendritic cell vaccination, median TTP was 19.9 months. Overall survival times in this group ranged from 18 to >58 months, with a median survival of 35.8 months. This compares favorably even when compared with historical/concurrent data for the best prognostic group of glioblastoma multiforme patients (recursive partitioning analysis class III: age under 50 years and Karnofsky performance score of ≥90) treated at UCLA during the same time period, who underwent surgical resection (not just biopsy) and became off steroids within 2 weeks after completion of postoperative radiotherapy (n = 99). Prins et al., 2011 was a dose escalation phase 1 study involving 23 patients, 15 of which had newly diagnosed glioblastoma and the other 8 were recurrent. The median OS time of all patients, taken from the date of initial surgical diagnosis of glioblastoma, was 31.4 months. OS from the time of initial diagnosis at 1, 2, and 3 years was 91%, 55%, and 47%, respectively. The median OS is 35.9 months (with a mean follow-up time of more than 4 years) in the 15 patients with ndGBM. Taken together these early trial though open-label and not controlled suggest potential efficacy for DCVax-L.
Before comparing the current blinded phase III survival data to other trials it is first worth taking into account the inclusion/exclusion criteria used in the DCVax-L phase III. This has a vital impact on the analysis since the DCVax-L phase III data is most reliably compared to other trials which used similar inclusion/exclusion criteria. If we do not take this into account then our analysis is likely biased with any improved survival results just the consequence of selecting the healthiest patients to be the in trial. In terms of median age, KPS score, male/female ratio this trial does not appear to be selective of patients with better prognosis compared to the other trials that we will inspect. However there are 2 features that do require deeper inquiry:
This is the one major inclusion/exclusion criteria that is expected to greatly improve the prognosis for the patients in the trial since patients with gross resection typically have a much better prognosis than those with only biopsies. 63% of patients in the DCVax-L phase 3 trial has a gross resection whilst 37% had at least a partial resection. No patients were biopsy only which is understandable since a minimal amount of tumour tissue is required to make the lysate.
In the DCVax-L trial any patient that had signs of progressive disease 2 weeks after the completion of chemoradiotherapy was no longer eligible to be part of the trial. This is a double-edge sword for the expected survival time for the patients in the trial since this results in the removal of typically longer living pseudoprogression patients (psPD - those that show signs of progression but which has reverted by the following scan) and the removal of the typically shorter living early progressors (ePD that have genuine progressive disease). The comparison to the brandes et al., 2008 data is particularly important in this case since they also removed ePD and psPD patients though 1 month after completion of chemoradiotherapy.
Now taking into account these inclusion/exclusion criteria it is worth comparing the DCVax-L blended results to that of other trials. There are 4 other trials run by other researchers that are worth comparing the blended data to: Stupps et al., 2017, Brandes et al., 2008, Lai et al., 2011 and Weller et al., 2017.
The Brandes study was a retrospective study but is a particularly good comparison for this trial since it has two important features relating to the inclusion/exclusion criteria. Because the aim of the brandes trial was to study the effect of promoter methylation and bisulphite sequencing to determine methylation status requires lots of DNA, this patient data set also weighed heavily towards patients with more resected tumour (only 1 patient of 101 was biopsy only). Furthermore, this trial separated patients based on having early progression, pseudoprogression and non progressive disease 1 month after the completion of chemoradiotherapy. This is similar to the DCVax-L trial which performed the same analysis 2-weeks after completion of chemoradiotherapy. The mOS of the non progressive disease patients from this trial came out at 20.2 months compared to 23.1 months for the blended DCVax-L phase III patients.
The Lai et al., 2011 study was a phase II trial of bevacizuab with SOC compared to SOC. It is useful since it splits the patients into recursive partitioning analysis (RPA) classes which have difference prognosis. Due to the inclusion/exclusion criteria the patients in the DVax-L phase III most likely resemble the patients from RPA class III/IV where patients are less than 50 years old or are over 50 but have a KPS score >70, partial/total resection and normal neurological function. Note there are likely to be some RPA class V patients in the DCVax-L trial so comparison solely to RPA class III/IV is conservative. The mOS of RPA class III/IV patients on BV/RT/TMZ from this trial came out at 20.8 months compared to 23.1 months for the blended DCVax-L phase III patients.
The Optune trial (Stupps et al., 2017) was a 695-patient open label phase 3 trial comparing the optune device (Tumour treating Fields) + SOC to SOC (randomized 2:1 - treatment+SOC:SOC). Because of the open label nature of this trial combined with the frequency of censors on the optune arm I believe this data is significantly biased. However, comparison to the SOC arm of this trial is meaningful. The SOC arm of this trial for the gross resection subset of patients came out with an mOS of 18.5 months compared to 23.1 months for the blended DCVax-L phase III patients.
Weller et al., 2017 was a trial of rindopepimut (Act IV trial), a vaccine targeting the EGFR deletion mutation EGFRvIII, consisting of an EGFRvIII-specific peptide conjugated to keyhole limpet haemocyanin. The control arm of this trial came out with an mOS of 20.0 months and rindopepimut arm of 20.1 months in the minimum residual disease subgroups of this trial (those most similar to gross resection) resulting in early termination for futility at a preplanned interim analysis. In total 745 patients were enrolled (405 with MRD, 338 with significant residual disease [SRD], and two unevaluable) and randomly assigned 1:1 between the control+SOC and rindopepimut+SOC arms.
Taken together therefore the blended data from the DCVax-L phase 3 look very good, outperforming other data sets with similar levels of gross resection. In summary the mOS of 23.1 months of the blended patients from thr DCVax-L phase III compare favorably to the 20,2, 20.8, 18.5 and 20.1 months mOS of the respective comparitor trial most equivalent patient subsets. For the following numerical analysis I have taken the reverse engineered surival data from the Brandes et al., 2008 and Lai et al., 2011 studies as the null hypothesis data (null hypothesis that the drug does not make any difference) as I believe the inclusion/exclusion criteria from these 2 studies are the closest to that of the DCVax-L phase III.
Here I have used previously published data of patients on Standard of Care (SOC) with similar exclusiion/inclusion citeria to model the NULL situation for the current phase III trial (See Analsyis tab for the simulation results). I reverse engineered the survival times off of the published kaplan-meier graphs hence there could be some errors. I have used 2 different data sets that I believe are the closest to the patient characteristics in the DCVax-L phase III based on inclusion/exclusion criteria. 1) That for Lai et al., 2011. For this data set I used the RPA class III/IV BV/RT/TMZ patient data (39 patient events and censors figure 3c) that should be equivalent to the patients selected for the DCVAx-L phase III. 2) The Brandes et al., 2008 non ePD or psPD patient data (35 patient events figure 3 orange line) which again selected patients in a similar manner to that of the DCVax-L phase III. Since the DCVax-L phase III removed patients with signs of progression 2 weeks after chemoradiochemotherapy whilst the Brandes study did the same thing after a month. Also these patients have a comparable level of resection to the patients in ther DCVax-L phase III trial with only a single patient out of the full 103 being biopsy only.